![]()
![]()
![]()
![]()
![]()
![]()
![]()
Running Gaussian in a Combined Cray PVP and T3E Environment
Carlos P. Sosa and John E. Carpenter
Chemistry Applications, Silicon Graphics, Inc./Cray Research, Eagan, MN 55121
cpsosa@cray.com and jec@cray.comwwwapps.cray.com/~cpsosa/ABSTRACT:
|
Running Gaussian in a Combined Cray PVP and T3E EnvironmentCarlos P. Sosa and John E. Carpenter Chemistry Applications, Silicon Graphics, Inc./Cray Research, Eagan, MN 55121 cpsosa@cray.com and jec@cray.comwwwapps.cray.com/~cpsosa/ABSTRACT: |
Users of ab initio quantum chemistry programs are attempting to understand the electronic structure of molecules[1]. The methodologies used are derived from very basic quantum mechanical theories and can be very accurate. However, this accuracy comes at a cost of rapidly increasing computer time, often nonlinearly, as the size of the molecule increases. A number of ab initio quantum chemistry programs exist, but the most widely used is Gaussian[2]. Because of the computational complexity of quantum chemical methods, users of Gaussian have always run on the fastest machine that they can find. It is for this reason that developers of quantum chemistry programs have considered parallelism to be the ultimate answer for getting accurate solutions in the shortest amount of time.
Starting with Enrico Clementi's efforts in the 1980's to the present day, a number of researchers have contributed to parallelization efforts on different quantum chemistry programs[3]. Parallelization first appeared in Gaussian 92 using shared memory constructs[4] and which was later extended to the Cray parallel vector (PVP) supercomputer family using directive based parallelism[5]. At roughly the same time, Gaussian, Inc. and Scientific Computing Associates combined efforts to develop a parallel version of Gaussian for distributed memory environments using the Linda programming model[6]. Recently, this version was ported to the Cray T3E supercomputer by chemists at Cray Research and Gaussian, Inc.[7]. The Cray T3E supercomputer differs from the Cray PVP supercomputer family in that it consists of many processing elements connected via a high speed network. Each processing element consists of a reduced instruction set (RISC) microprocessor and memory.
Many supercomputer centers around the world have Cray PVP supercomputers like the Cray C90, Cray T90 and Cray J90 supercomputers that run a large number of Gaussian calculations many of which take long hours to complete. Some of these centers have installed Cray T3E supercomputers as their next generation supercomputer. Given that Gaussian versions now exist for both Cray PVP and Cray T3E supercomputers, there naturally arises the question of which platform is best suited for running the program. In general, Gaussian takes into account resources available at run time and chooses the best algorithm for a particular single hardware configuration. See ref.[8] for resources used by method and job type. However, Gaussian cannot determine which machine it should be run on to begin with. It is the purpose of this paper to make recommendations about which Gaussian jobs can be efficiently done on the Cray T3E and which jobs are best done on the Cray PVP platforms. Given this information, it may be possible to develop a mechanism that would actually choose the best computer platform in a computer environment to run a given Gaussian calculation. This may be implemented in a queueing system such as the network queueing environment (NQE).
In the following sections, we will more precisely describe the Cray PVP and T3E supercomputers, describe which features of Gaussian are parallelized, detail the jobs we used to measure the performance of the two Cray PVP and T3E supercomputers, give the performance information, and finally, use that information to develop a first level recommendation of where Gaussian should be run most efficiently.
Three supercomputers were used in this study, two PVP machines and one MPP. The PVP machines correspond to Cray J916 and Cray T94. The MPP machine used in this study is the Cray T3E. The Cray J916 is an entry-level PVP machine. It has a clock period of approximately 10 ns. The peak performance per processor approaches 200 MFLOP/s. Memory technology is based on CMOS DRAM with a memory access of 70 ns. The Cray J90 has parallel vector processing capabilities. Each processor in the Cray J916 series has a cache for increased scalar performance. This cache complements the systems' shared memory bandwidth.
The Cray T94 supercomputer has high-speed processors, each with a peak performance of approximately 2 billion floating point operations per second (2 GFLOP/s). The Cray T94 system provides up to 4 processors, 1024 Mbytes of central memory. Each memory stack contains 40 4-Mbit SRAMs for a total of 2 million 64-bit words. There are 16 stacks on a memory module.
The Cray T3E parallel computer system consists up to 2048 process elements (PEs) and 128 system/redundant PEs. Peak performance for the largest configuration is 1 TFLOPS. Each PE on this system is composed of a DEC Alpha 21164 EV5 RISC microprocessor. Memory size scales from 64 MBytes to 512 MBytes per PE (it can be extended to 2 GBytes). The interconnect network on the Cray T3E is a 3D-torus with some partial planes allowed. Process elements are built 4 to a printed circuit board. These 4 PEs have network connection to a globally-accessible I/O channel. Finally, it is important to mention that the EV5 has a 96 KBytes 3-way set associative secondary cache on chip [9].
Gaussian [2], a connected series of programs can be used for performing a variety of semi-empirical, ab initio, and density functional theory calculations. Each program communicates with each other through disk files. All the individual programs are referred as links, where links are grouped into overlays [10]. The Gaussian architecture on shared or distributed memory machines is basically the same, that is, each link is responsible for continuing the sequence of links by invoking the exec() system call to run the next link. Memory is allocated dynamically. It is important to note that on distributed-memory versions this amount of memory is allocated per processor.
Parallelization of Gaussian on PVP and MPP machines has been achieved differently. On PVP systems compiler directives are currently being used [5]. Directives distribute the work among the processors at the DO LOOP or subroutine level. On the other hand, parallelization of Gaussian on the Cray T3E relies on Linda [7]. Linda is a model for parallel processing based on distributed data structures [11]. Processors and data objects communicate by means of the tuple space (TS) or Linda memory. The interaction between the tuple space and the processors is carried out by four basic operations: out, in, eval and rd. These operations add/remove data objects from/to the tuple space. eval on the other hand also forks processes to initiate parallel execution. In both implementations parallelization has been carried out in the computation of the two-electron repulsion integrals via the Prism algorithm[12]. Since the Prism algorithm is called from several links, effectively, several links have been parallelized using this scheme.
In terms of functionality there are two basic differences between PVP and MPP machines. In the currently available version, MP2 has been parallelized in the Linda or T3E version, but not in the PVP version. On the other hand, MPn/QCI methods can take advantage of multitasked library routines to leverage multiple-processors on vector machines.
Table I shows the options that can take advantage of multiple-processors on different architectures.
Table I. Levels of theory that can be done in parallel on either or both parallel vector (PVP) or massively parallel (MPP) supercomputer architectures.
| Level | Single-point | Optimization | Frequency |
| Hartree-Fock | PVP, MPP | PVP, MPP | PVP, MPP |
| Density Functional Theory | PVP, MPP | PVP, MPP | PVP, MPP |
| CI-Singles | PVP, MPP | PVP, MPP | PVP, MPP |
| MCSCF | PVP, MPP | --- | --- |
| MP2 | MPP | MPP | MPP |
| MP3 and MP4 | PVP | --- | --- |
| QCI and CC | PVP | --- | --- |
There exist hundreds of options for running Gaussian, but there are three major characteristics of a Gaussian job which determine the computational complexity of the calculation: job type, theoretical method, and basis set size. The basic job type is to calculate the total energy of a molecule. Furthermore, one can calculate the first and second derivatives of this energy with respect to the positions of the atoms in the molecule or with respect to things like an external electric field. This functionality can used to optimize molecular geometries and compute the normal modes and frequencies of vibration of a molecule. Owing to the importance of molecular structure to chemistry, a large majority of calculations run at supercomputer centers are geometry optimizations followed by frequency calculations. Another type of calculation that is less often run, but holds value because of its accuracy are single point energy calculations at high levels of theory. Figure I. gives the relative usage of these different theoretical methods at a typical supercomputer center and shows the preponderance of geometry optimization in Gaussian usage at these sites.
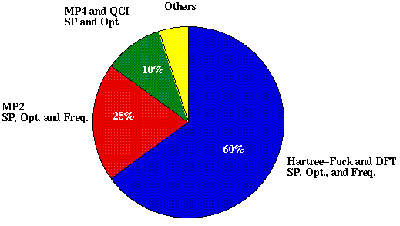
Figure I. Estimated Gaussian usage at any given computer center.
We have chosen to use single point energy (SP), FORCE (the time required for a geometry optimization is a multiple of the time needed for a FORCE calculation) and FREQ calculations in this study as a representative set of job types being run in supercomputer centers. While it is known how to solve the time independent Schrodinger equation in the Born-Oppenheimer approximation exactly, it is not practical to do so for all but a few small molecules due to huge computational complexity. A large number of approximate theoretical methods that range in accuracy and computational cost have been developed, and Gaussian is capable of doing most. Without discussing each theoretical method in detail (see, for example, [1]), we have chosen to use Hartree-Fock (HF), the three parameter density functional method due to Becke [8] (B3-LYP), and second and fourth order Moeller-Plesset theories (MP2 and MP4) in our study. In order of accuracy and computational cost these computational methods go in increasing order: HF < B3-LYP < MP2 < MP4). The HF and B3-LYP methods are widely used to optimize geometries while the MP2 and MP4 methods are more often, but not exclusively, used to calculate energetics after optimization (see Figure I). This is why we have chosen to use these methods over others. Finally, the size of the basis set is dictated by the number and type of atoms in the molecule as well as the precise basis set used. In general, the number of basis functions is a linear function of the number of atoms. Gaussian contains a large number of internal basis sets and has the ability to read in a basis set. For a more complete description of basis sets, see, for example, [1,8]. Given choices for the job type, theoretical method, molecule and basis set, we generated seven jobs that were used to measure performance of Gaussian running on the Cray PVP and Cray T3E supercomputers. These are detailed in Table II.
Table II. Gaussian jobs generated to measure the performance of Cray PVP and T3E supercomputers.
| Job | Job Type & Basis Set | Molecule |
|---|---|---|
| Job I | FORCE CIS/6-31++G | acetyl-phenol |
| Job II | SP MP4/6-31G(3d,3p) | methylhydrazine |
| Job III | SP HF/6-311G(df,p) | 2-pinene |
| Job IV | SP B3-LYP/6-311G(df,p) | 2-pinene |
| Job V | FORCE B3-LYP/6-311G(df,p) | 2-pinene |
| Job VI | FREQ B3-LYP/6-311G(df,p) | 2-pinene |
| Job VII | SP MP2/6-31G* | eicosane |
The calculations were carried out on 1 processor of the Cray T90, 1, 2 and 4 processors of the Cray J90 and 1, 2, 4, 8, 16, 32 and 64 processors of the Cray T3E. This is perhaps the most controversial choice of the study because, in principle, we could run on as many as 32 processors of the Cray T90 and J90 supercomputers. However, Cray T90 supercomputers are often used by large numbers of researchers at a given time and the use of parallelism at these sites is difficult. This is less true about Cray J90 usage and so we have chosen to use a small number of processors in parallel on that machine.
Job I. CIS/6-31++G FORCE calculation on acetyl-phenol. One key difference between PVP and MPP machines is in the memory architecture. The current version of Gaussian takes advantage of the shared-memory on PVP systems. Figure II shows a speedup of 8X for an incore calculation (CIS/6-31++G FORCE SCF=INCORE) when compared to the same calculation using a direct algorithm. In general, incore calculations tend to show speedups between 2X to 8X versus direct algorithms. This case consists of a total of 154 basis functions, it would require to have approximately 80MW per PE on the Cray T3E. In order to take full advantage of the memory on the T3E, it is required to distribute memory allocation among all the processors. This feature will be developed in future releases of the program.
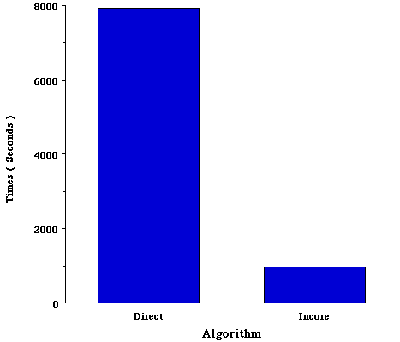
Figure II. The time required for a incore CIS/6-31++G calculation on acetyl-phenol carried out on a single processor of a Cray J916 Supercomputer.
Job II. MP4/6-31G(3d,3p) SP calculation on methylhydrazine. Another set of options that show an advantage (using Rev E.2) by running them on PVP machines over MPPs correspond to highly-correlated methods. Figure III illustrates, first of all the speed of a single processor on all the machines, it indicates that for this calculation, single processor T3E is roughly equivalent to single processor J90. However, the most important point when using this version of the program, is that this case can use multiple processors on vector machines by leveraging highly-optimized Cray math libraries. We are currently working on the parallelization of post-Hartree-Fock methods on the T3E.
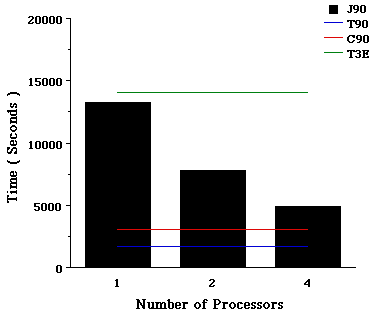
Figure III. Time required for a MP4SDTQ/6-31G(3d,3p) SP calculation on methylhydrazine using Cray T90, C90, J90, and T3E supercomputers.
Jobs III and IV. HF and B3-LYP/6-311G(df,p) SP calculations on 2-pinene. Figures IV and V compares the performance of 2-pinene, Hartree-Fock and DFT on the T3E, J90, and T90. Clearly, single-processor performance on the T3E is approximately 3X slower when compared to single-processor performance on the J90. This large difference in single-processor performance may be overcome by using the newest release of Cray compilers (available in programming environment 3.0). In terms of scalability, for systems like 2-pinene, Hartree-Fock and DFT show a scalability of 32 to 37 processors out of 64. In this particular case, 99.5% of the time is spent in the SCF iterative scheme (L502).
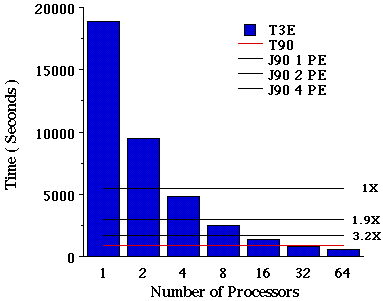
Figure IV. Time required for a RHF/6-311G(df,p) single-point energy calculation on 2-pinene using Cray T90, J90, and T3E Supercomputers.
Thus, little overhead is introduced by the fact that some of the links are running sequentially. As the number of processors is increased, the amount of time spent computing the two-electron integrals decreases and the sequential diagonalization of the Fock matrix starts decreasing the parallel efficiency[7]. On the other hand, increasing the size of the basis sets for the same system (2-pinene), the scaling is almost linear up to 128 process elements[7].
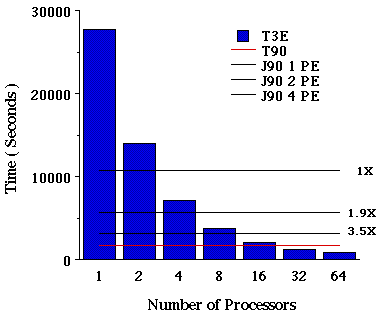
Figure V. Time required for a B3-LYP/6-311G(df,p) single-point energy calculation on 2-pinene using Cray T90, J90, and T3E Supercomputers.
Jobs V and VI. B3-LYP/6-311G(df,p) FORCE and FREQ calculations on 2-pinene. Figure VI and VII summarizes elapsed timings and speedups for a gradient and frequency calculations, respectively. These cases correspond to a B3-LYP calculation on 2-pinene. The total B3-LYP energy is an eigenvalue problem. The two-electron integrals contributing to the B3-LYP energy can be computed by means of the Prism algorithm[12].
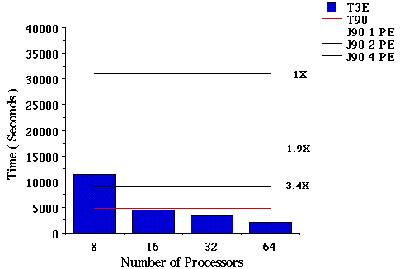
Figure VI. Time required for a B3-LYP/6-311G(df,p) gradient calculation on 2-pinene using Cray T90, J90, and T3E Supercomputers. Note that long computation time prevented us from doing the calculation on 1, 2, and 4 processors of the Cray T3E Supercomputer.
In general, we see that DFT calculations with first and second derivatives are well suited for the T3E. With the current version of the program, 8 to 16 processors give performance equivalent to 1 processor on the T90. Again, this ratio will decrease with the new generation of compilers.
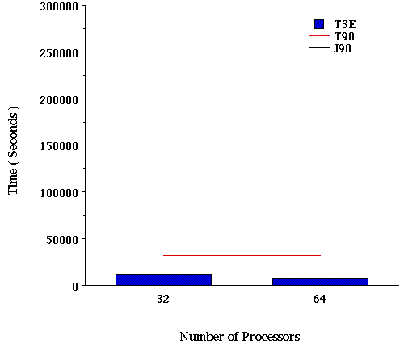
Figure VII. Time required for a B3-LYP/6-311G(df,p) frequency calculation on 2-pinene using Cray T90, J90, and T3E Supercomputers. The long time required for this calculation prevented us from doing the job on 1, 2, 4, 8, and 16 processors of the Cray T3E supercomputer.
Job VII. MP2/6-31G(d) SP calculation on eicosane. Finally, a single-point energy calculations was carried out at the MP2 level of theory. In the case of the MP2 calculation, increasing the number of PEs decreases the number of integrals that are computed per PE (or worker). In the case of 1 PE we have a total of 6 batches of integrals, a total of 3 batches are generated for 2 PEs, 2 batches for 4 PEs and finally only 1 batch of integrals for more than 4 PEs. Batches of integrals are groups of integrals based on a common molecular orbital index that can be computed at the same time[7]. Figure VIII illustrates the performance of parallel MP2 using a semi-direct algorithm. This data illustrates an almost linear speedup up to 8 process elements on a T3E. Currently this option cannot take advantage of multi-processors on a PVP machine.
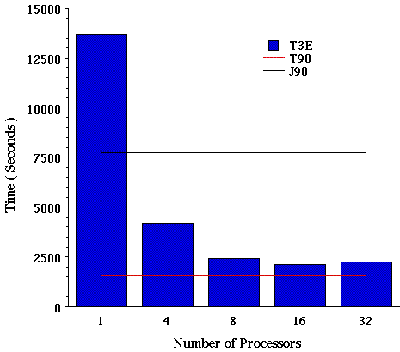
Figure VIII. Time required for a MP2/6-31G(d) single point energy calculation on eicosane using Cray T90, J90, and T3E Supercomputers.
SCF and DFT calculations have been shown to run efficiently on Cray T3E systems using up to 32 and 64 process elements. However, scaling is clearly dependent on the number of basis functions and the size of the molecule. In this study we have looked at 2-pinene as an example of a molecule that shows good scaling. Further investigation will be carried out for larger systems where cutoffs may play an important role in the number of two-electron integrals that are performed per process element. It has also been shown that single-processor performance on the Cray T3E is slower than expected. This is due to the fact that additional compiler optimization is required to improve single-processor performance. In general, the results show that using 16 or 32 processors of the Cray T3E is an effective way to do SCF and DFT calculations more rapidly and jobs of this type should be steered toward the Cray T3E supercomputer.
The CI-Singles approximation provides a direct method of computing excited states with minimal disk storage required. Due to its simplicity, CI-Singles provides a good approximation for large systems. In addition, in this study we have shown that this technique is well suited for highly-parallel machines. On the other hand, incore calculations tend to favor PVP machines since total memory on the Cray T3E is allocated per processor. This implies that relatively small CI-Singles calculations should be steered toward large memory Cray PVP machines while larger CI-Singles calculations should be done on 16 or 32 processors of the Cray T3E supercomputer.
MP2 calculations on the other hand are more I/O demanding. In the case of MP2 calculations the scaling is closely coupled to the number of batches distributed among all the processors. In the case presented in this study a semi-direct algorithm was used. This algorithm is disk-based, and therefore most of the process elements are involved in carrying out I/O. The Cray T3E contains a large number of I/O channels, one per PE module ( the T3D contained a small number of dedicated, high-bandwidth I/O nodes). We do not expect a very large overhead for this particular case. Because the Linda version of Gaussian does MP2 calculations in parallel while the PVP does not, it is reasonable to steer moderate size MP2 calculations to 16 to 32 processors of the Cray T3E supercomputer while leaving large MP2 calculations on the Cray T90 machine. This needs more investigation.
Finally, highly accurate and computationally demanding MP4 calculations should be done on Cray PVP machines due to the the ability to use multiple vector processors. The Linda version of Gaussian 94 does not yet do MP4 calculations in parallel. We will be working on that in the near future.
The authors wish to thank the Corporate Computing Network (CCN) at Silicon Graphics, Inc./Cray Research for providing all the hardware to carry out this study. We also thank G. Scalmani for valuable discussions and providing us with a copy of the poster that he presented at the III National Conference of computer science and chemistry.
1 Gaussian is a Gaussian, Inc. trademark.
Carlos Sosa is a computational chemist at Silicon Graphics, Inc./Cray Research in Eagan. He currently works with Gausian 94 on Cray Research supercomputers.

cpsosa@cray.com
http://wwwapps.cray.com/~cpsosa/
John Carpenter is manager of the chemistry group at Silicon Graphics, Inc./Cray Research in Eagan.

jec@cray.com